
AI in Drug Discovery - Please Stop Fishing in the Bathtub!
Published:

The Hype Cycle and the Reality of Virtual Screening
Working on machine learning in drug discovery can be profoundly frustrating. Every week, the purveyors of social media hype herald a new “revolution” in AI for the field. As someone deeply invested in this space, I often follow the trail to these revolutionary papers, only to find researchers repeating the same fundamental mistakes that have plagued the discipline for more than a decade.
Much of the recent hype has centered on virtual screening. Some recent papers have touted approaches that purportedly enable screening of the entire proteome in mere hours. These results appear impressive—until you realize they are validated against datasets that were effectively discredited seven years ago. A primary offender, and one I will focus on here, is the DUD-E dataset. Originally developed to validate docking methods, it has been frequently (and incorrectly) used to validate machine learning models.
What is the DUD-E Dataset?
In the early days of virtual screening (the late 1980s and early 1990s), researchers validated their methods by “seeding” known active compounds among inactive decoy structures. The goal was to determine whether a virtual screening method—typically docking—could successfully separate the two. Although the logic appeared sound, many early test cases were unduly easy because of inherent physical differences between the actives and the decoys.
Consider the first-generation HIV protease inhibitors. These were large molecules; Saquinavir has a molecular weight of 671, and Amprenavir is 506. If these structures are “hidden” among a set of decoys with molecular weights in the 350–450 range, any algorithm—especially docking, which is biased to reward larger molecules with higher scores—can “cheat” by simply picking out the biggest structures.
To address this bias, John Irwin, Brian Shoichet, and their colleagues developed the Directory of Useful Decoys (DUD). They collected active compounds and designed decoy sets whose physical properties matched those of the actives. By aligning these distributions, the authors hoped to eliminate size and property bias from the validation process. In addition to molecular weight, they matched lipophilicity (LogP), charge, and the number of hydrogen-bond donors and acceptors. This collection evolved into DUD-E, an enhanced collection of activities and decoys that incorporated topological similarity and property distributions when constructing validation sets.
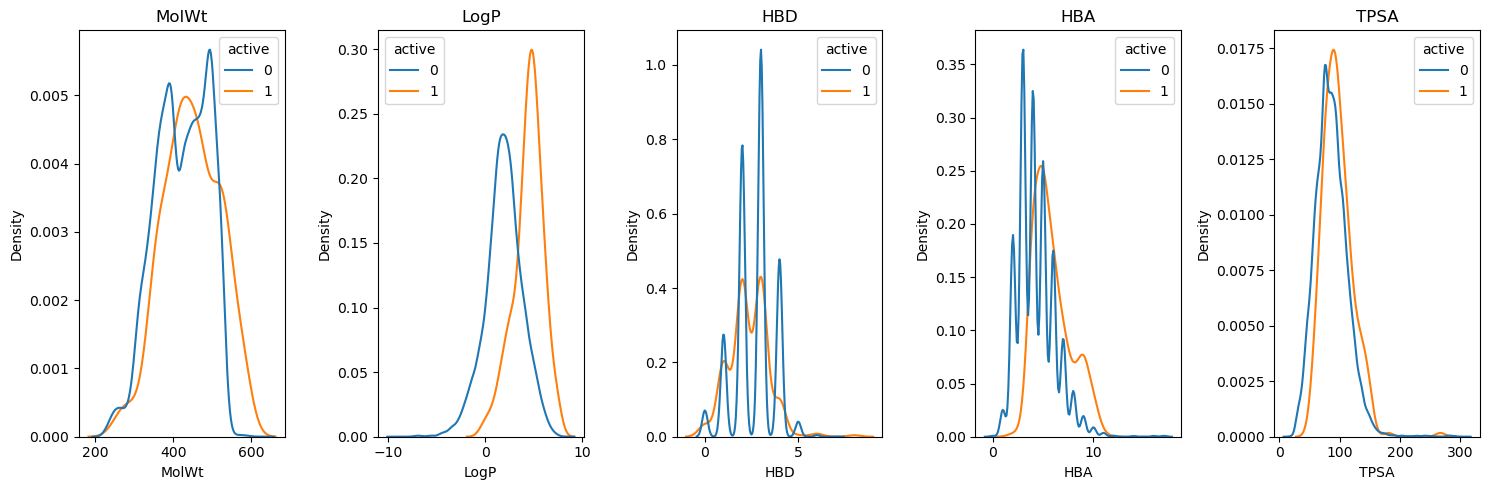
For example, consider the AKT1 active and decoy subset of the DUD-E dataset. The figure below shows the distributions of five key physical properties for active and decoy compounds. The sets have nearly identical molecular weight and polar surface area distributions. The differences in the LogP distributions are likely due to my use of a different LogP implementation than the one used to generate the DUD-E set.
The Fatal Flaw: Chemical Dissimilarity
If the physical properties are matched, what’s the problem? While DUD-E balances bulk properties like molecular weight and LogP among the actives and decoys, the decoys were intentionally designed to be structurally distinct from the actives. This was done to ensure that the decoys were truly inactive. While this makes DUD-E an excellent benchmark for docking, it makes it a poor choice for machine learning.
In fact, the DUD-E authors point this out explicitly in their paper: “Fundamentally, DUD and DUD-E are designed to measure value-added screening performance of 3-D methods over simple 1-D molecular properties. Decoys that might bind are removed using 2-D ligand similarity, so DUD-E is inappropriate to test 2-D methods.”
To visualize why the DUD-E set is inappropriate for ML, I used t-SNE (t-distributed Stochastic Neighbor Embedding) with RDKit Morgan Fingerprints to map the chemical diversity across six DUD-E datasets. In this projection, each point represents a molecule; similar structures cluster together, while dissimilar ones are pushed apart.
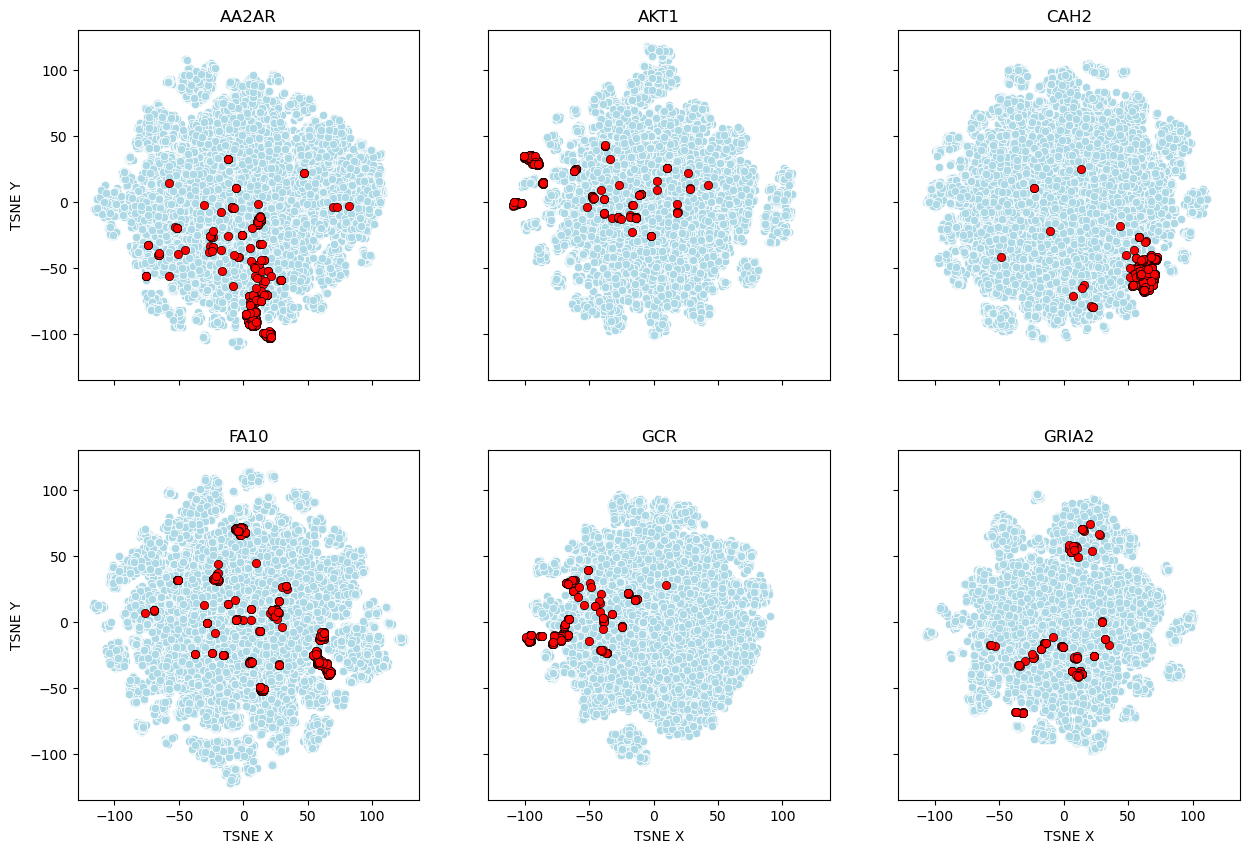
The result is striking. In the maps above, the active compounds (red) form tight, isolated clusters, while the decoys (light blue) are scattered elsewhere. This reveals the “analog bias” inherent in the dataset. Because the actives are often structurally related to one another but completely distinct from the decoys, a machine learning model doesn’t actually have to learn anything about protein-ligand interactions. It only has to learn how to recognize the “red” clusters.
It doesn’t take a sophisticated deep-learning architecture to “solve” DUD-E. In the next section, I’ll show how easily a simple, baseline ML model can identify the actives with near-perfect accuracy—further proving that we aren’t measuring model performance but rather a flaw in the data.
This Is Not News
What I’m pointing out here is not new information. Many of the issues with the DUD-E dataset and ML models were reported in a 2019 paper by Tom Kurtzman, David Koes, and colleagues. In this paper, the authors evaluated the ability of Convolutional Neural Networks (CNNs) to identify active compounds in the DUD-E datasets.
Kurtzman and coworkers found that their models were equally predictive when they “turned off” the protein representation. In fact, there was a near-perfect correlation between the affinities calculated with and without the protein. Several additional experiments showed that the CNN wasn’t learning from the protein and was instead simply learning to distinguish active molecules from decoys. They demonstrated that one could train a model on complexes from a different protein and still distinguish actives from decoys.
A Simple Demonstration
To illustrate the flawed nature of the DUD-E dataset for ML model validation, we will train a model on each of the 28 kinase datasets in the DUD-E set and examine the ability of that model to separate the actives from the decoys in the other kinase datasets.
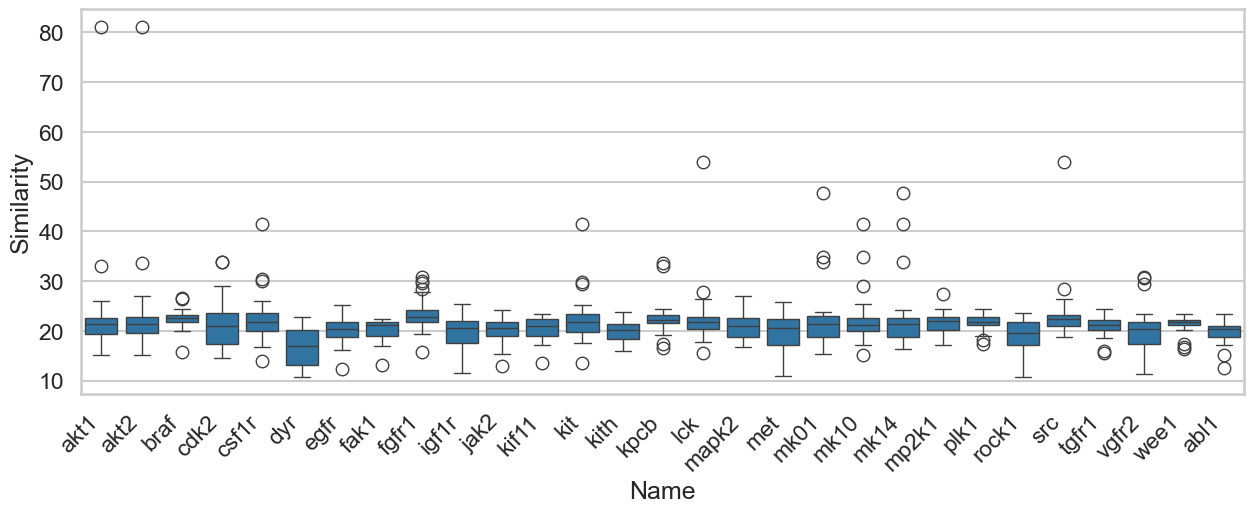
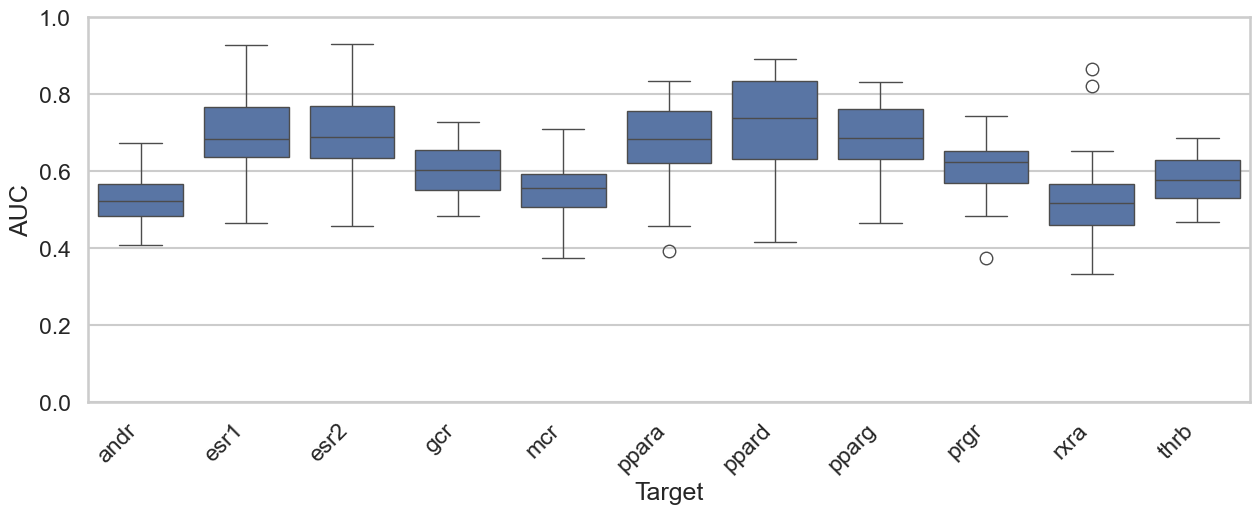
Before we begin, we should address the elephant in the room. Many will point out that since these targets are all kinases, this is an easy task. When building models to predict protein-ligand binding affinities, most groups use a 90% overall sequence similarity cutoff to separate their training and test sets. I’m not arguing this is the right way to do it; I’m simply pointing out what people do. As shown in the box plot below, according to conventional criteria, none of these targets would be considered “similar.” In fact, the only pair approaching the 90% cutoff is AKT-1 and AKT-2.
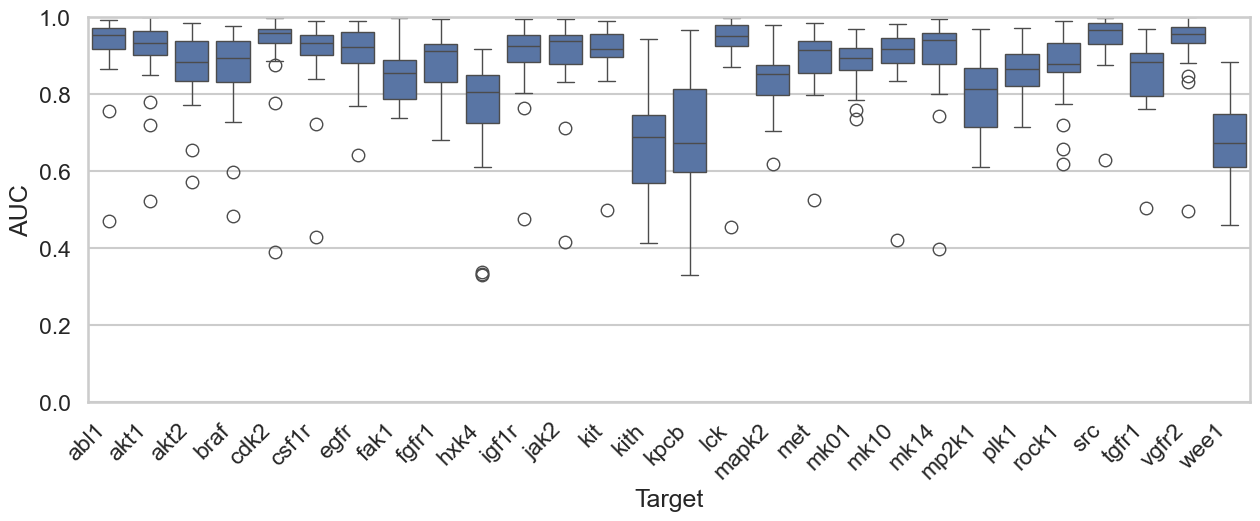
With the obvious out of the way, let’s build some models. I employed a simple strategy that has become my initial “go to” for ML model building: I generated 2048-bit Morgan fingerprints with radius 2 for the molecules in the 23 DUD-E kinase datasets. I then used these fingerprints to train a LightGBM classification model using each target as a training set and predicting on all of the other datasets.
The results are shown in the figure below. We have the 28 DUD-E kinase datasets on the horizontal axis and the AUC (Area Under the Receiver Operating Characteristic) on the vertical axis. We observe that, in most cases, a model trained on data for a single kinase can readily distinguish active molecules from decoys across all other kinases.
To push this silly test even further, we can examine whether a model trained on kinase data can separate active and decoy molecules from the DUD-E nuclear receptor targets. As shown in the plot below, the models trained on kinase data showed weak predictive ability when tested on nuclear receptor actives and decoys, but did not perform as well as when tested on kinase targets. Given the lack of similarity between kinase inhibitors and molecules that bind to nuclear receptors, this isn’t unexpected.
Some Closing Thoughts
While reading recent papers, I was surprised to find that they used the DUD-E dataset as a validation set. As I noted above, many authors have pointed out that this dataset is inappropriate for ML. If we have a model trained on thousands of datapoints from ChEMBL and other databases, tasks such as classifying molecules from the DUD-E dataset become trivial.
How did we get here? How do we have papers published in high-profile journals that make bold claims based on incredibly weak evidence? I don’t believe that people are being intentionally dishonest; they are simply failing to familiarize themselves with prior work done in the field. This is a prime example of those who do not study history being doomed to repeat it. As new groups come into drug discovery from computer science and related fields, it’s important for them to read the literature and familiarize themselves with work that came before.
Unfortunately, our field is littered with poorly constructed benchmarks that later become “standards” for comparison. I believe that journals need to assume greater responsibility for what they publish by defining clear standards and criteria and by using editors and reviewers with deep expertise in the field.
One final note to editors and reviewers who might be reading this: I commend journals for establishing guidelines that require including source code with a paper. However, when I look at the code repository for a recently published paper and see that the last commit was more than two years ago, the chances that I’ll be able to run that code are essentially zero. In the fast-moving world of machine learning, two years is an eon. It’s always good to make an effort to ensure the code is up to date and runnable. The same goes for authors. You’re releasing your code so that people can use it and build upon it. Make that easy to do.
If anyone is interested in reproducing or extending this work, all code is available on GitHub..




{kind=link}